Research
My research focus is algorithm development for sequence
analysis, in particular
- detection and analysis of DNA repeats and
- analysis of multiple sequence alignments (both protein
and DNA) for functional and structural information.
Tandem Repeats. A tandem repeat
is an occurrence of two or more adjacent, often approximate
copies of a sequence of nucleotides. As an example, consider the following
visualization of a tandem repeat taken from the Tandem Repeats Database (TRDB). The blue line
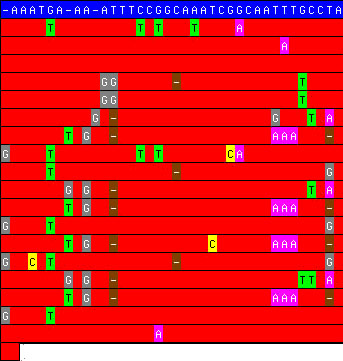 is the consensus pattern and the
19 red lines are
the individual copies within the repeat, one copy per line. Only
differences with respect to the pattern are shown. Note the redundant
mutations which suggest that this repeat has undergone several rounds of
expansion. is the consensus pattern and the
19 red lines are
the individual copies within the repeat, one copy per line. Only
differences with respect to the pattern are shown. Note the redundant
mutations which suggest that this repeat has undergone several rounds of
expansion.
Tandem repeats are ubiquitous sequence features in both prokaryotic and
eukaryotic genomes. In humans, they are known to cause at least ten inherited
neurological diseases including fragile-X mental retardation, Huntington's
disease, and myotonic dystrophy and are associated with a number of other major
illnesses, including diabetes, epilepsy, and ovarian and other cancers.
Additionally, they are the basis of DNA fingerprinting and have recently been
used to discriminate between different bacterial strains, including
anthrax strains.
Detecting approximate tandem repeats had been a difficult open problem.
Pattern sizes vary from a few nucleotides to well over 1000 and conservation
between the copies can be lower than 70 percent. In order to address this need,
I developed the Tandem Repeats
Finder which employs a stochastic model of tandem repeats and
associated statistical detection criteria. It is extremely fast and thorough,
and is now regularly used to analyze new genomic sequences.
TRDB. Tandem repeats remain a neglected class despite their
ubiquity and known functional roles. Information sources are incomplete,
fragmented and difficult for researchers to access. The Tandem Repeats Finder
has simplified the task of identifying repeats (for example, analysis
of C. elegans with a genome of approximately 100 million bases takes 60
minutes and finds ~25,000 tandem repeats) opening the way for more detailed
analysis. I am in the early stages of building a multi-genome database (TRDB) of
tandem repeats. Ongoing work includes: 1) clustering repeats into families, 2)
developing sequence based predictive criteria for copy number polymorphism, 3)
automating the annotation of repeats and families, and 4) implementing user
interface and data visualization tools.
Other Repeats. I am working on methods to detect low-copy
repeats, which occur as non-adjacent copies, and composition repeats which occur
as variations in DNA composition rather than as `word' patterns. The former
contribute to many types of disease and the latter may have regulatory and/or
structural effects.
Extracting information from multiple sequence alignments.
Alignment of many related sequences from viral or bacterial quasi-species can
reveal important information about proteins, RNA, and DNA, including changes
that correlate with pathogenicity, drug susceptibility and sequence structure.
Extracting this information, manually, from multiple alignments is often
difficult, especially when a large number of long sequences are utilized. In
collaboration with researchers at Mount Sinai, I have been developing Mutation
Master a set of computer analysis tools which rapidly provide a
visual display and tabulation of site, frequency, number and likelihood of point
mutations. Analysis of hepatitus C virus (HCV) protein sequences using Mutation
Master has identified possible sites of amino acid structural interaction, and
has revealed that ARFP, a novel protein encoded in an overlapping reading frame,
is as conserved as conventional HCV proteins. Ongoing work includes developing
similar tools to analyze RNA multiple alignments for structural clues including
compensatory mutations.
Education
I have been teaching for 20 years, the first 8 as a high school mathematics
teacher and the remainder as a graduate teaching assistant, postdoc and
assistant professor in computer science. My area of expertise and main interest
in teaching is theoretical computer science/algorithms with applications in
biology, i.e., computational biology and bioinformatics.
While at Mount Sinai, I have developed and taught graduate level courses with
a focus on biological sequence analysis. These include Computational
Structural Biology and Advanced Topics in Computational
Molecular Biology. Topics covered in the advanced course include: 1)
sequence alignment algorithms, 2) scoring functions and substitution matrices
for alignment, 3) database search algorithms (BLAST and FASTA), 4) multiple
alignment, 5) hidden Markov models and their application to gene detection, and
6) algorithms for finding unknown repetitive patterns in sequences (enumeration,
Monte Carlo and statistical methods). I am currently developing another course
Pattern Detection Techniques for Biological Sequences which
incorporates topics in pattern detection for sequences as well as methods for
expression array analysis.
I have served as advisor/mentor for graduate students and have hosted several
undergraduate and high school students in my lab during summers at Mount Sinai.
I participated in NSF Young Scholars programs as a guest lecturer and served as
co-director of the Villanova Summer Research Institute in Biology, Computing
and Mathematics. I am currently collaborating on the development of high school
curriculum modules related to computational biology as part of the Gateway to
Higher Education program in New York City.
I support outreach efforts to attract and train minority students and
underrepresented groups and have actively participated in such activities
through the Mount Sinai summer fellowship program, the Mount Sinai Hospital
Placement program for high school students and the New York City Alliance for
Minority Participation in Science Engineering and Mathematics.
|







 Composition Alignment:
Composition Alignment: